
溴代烷
溴代烷,又名溴代烷烃,是有机溴化合物的一类,指的是烷烃分子中的一个或多个氢原子被溴取代的有机化合物,属于卤代烃的一种。通式为。而最简单的溴代烷为一溴甲烷(甲基溴)。它们的密度大都比水大,而同一烃基的卤代烷的密度顺序为氯代烷<溴代烷<碘代烷[1]。大部分的溴代烷都不溶于水、溶于大部分的有机溶剂[2]。


溴代烷可以用作有机溶剂和有机合成的中间体(烷基化试剂[3])。但溴代烷的毒性限制了其应用范围。而人类较常使用甲基溴做熏蒸的用途[4],但已渐渐被淘汰。
溴代烷天然存在于大自然中。据估计,海洋每年排放1到2百万吨的溴仿和56,000吨的溴甲烷[5]。红藻会把溴代烷和碘代烷浓缩在囊泡细胞里,紫杉状海门冬的挥发性精油中有95%都是卤代烃,其中有80%都是溴仿[6] 。下图是大自然中的有机溴化合物的例子,最左边的一种属于溴代烷:

左到右:溴仿、一种溴代二酚、骨螺紫、tambjamine B。

历史[编辑]
卤代烷在很久以前已被应用,如在1847年苏格兰医生詹姆斯·杨·辛普森及其朋友发现氯仿可以用作麻醉剂[7][8]。在溴代烷的结构方面,于1857年凯富勒发现碳有四价[9]、1861年布特列洛夫提出化学结构理论、1974年范霍夫和勒·贝尔提出碳的正四面体构型假说(又称范托浩夫—勒·贝尔模型),这些都对化学家对溴代烷结构的研究起了重大的作用。在合成溴代烷的方面,在19世纪,由于有机化学蓬勃而有系统的发展,化学家们找到更多合成溴代烷的方法[10]。例如俄国化学家亚历山大·波菲里耶维奇·鲍罗丁在1861年尝试用乙酸银来制备甲基溴时,发现了汉斯狄克反应。对于由烯烃合成卤代烷的反应(烯烃与氢卤酸的亲电取代反应)的区域选择性,在1869年,俄国化学家马可尼可夫发现亲电试剂中的正电基团(如氢)总是加在取代最少的碳原子上[11],但烯烃和溴化氢的反应却不一定得到这个结果。直到1933年莫里斯·S·卡拉施给出了解析:过氧化物效应[12],即是在过氧化物存在下,烯烃和溴化氢的反应是反马氏规则的,因为反应涉及到自由基。
分类[编辑]
跟其他卤代烷一样,溴代烷亦可分为一级(伯)、二级(仲)、及三级(叔)溴代烷。一般来说,连着溴原子的碳连接着多少个碳,就是几级溴代烷。
-
 一级溴代烷的结构
一级溴代烷的结构 -
 二级溴代烷的结构
二级溴代烷的结构 -
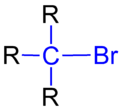 三级溴代烷的结构
三级溴代烷的结构


例如,1-溴丙烷为一级溴代烷,2-溴丙烷为二级溴代烷[13],2-溴-2-甲基丙烷为三级溴代烷。
命名[编辑]
溴代烷通常以普通命名法来命名,找出相应烷烃作为母体,将溴视作取代基,并以数字标明其位置(先写溴,再写烷基取代基,最后写母体)[14]。而不少溴代烃都有俗名,例如溴仿(三溴甲烷)。同时,亦可以把烷基当作是取代基来命名,即先写烷基再写溴,例如乙基溴、甲基溴等[15]。
合成[编辑]
烷烃的自由基取代反应[编辑]
烷烃在紫外光照的条件下,可以与溴反应生成相应的溴代烃和溴化氢。反应机理有三个步骤[16]:
- 键引发:
- 键增长:
- 一、溴自由基跟一分子烷烃反应,生成溴化氢和相应的烷烃自由基。
- 二、烷烃自由基跟另一分子的溴反应,生成溴代烷和另一溴自由基。
- 键终止:有三个终止的反应,直到自由基消失,反应结束。
- 1.两个溴自由基反应,生成一分子溴。
- 2.两个烷基自由基反应,生成另一碳链更长的烷烃。
- 3.一个溴自由基和一个烷基自由基反应,生成溴代烷。
反应是个连锁反应,会继续生成二溴烷烃、三溴烷烃、四溴烷烃[18]。
烯烃、炔烃的加成反应[编辑]
亲电加成[编辑]
烯烃和炔烃都具备π电子,可以与亲电试剂发生亲电加成反应。其中与溴化氢和溴的加成反应可制备溴代烷。这些反应一般遵从马氏规则。
烯烃、炔烃与溴完全反应,分别生成1,2-二溴代烷和1,1,2,2-四溴代烷。反应跟从环𬭩离子机理[19],如下图所示。其中在溴分子接近烯烃、炔烃时,形成了诱发偶极,受到π电子的进攻,形成溴𬭩离子。此溴𬭩离子再受到带负电荷的溴离子的进攻,生成产物。

烯烃、炔烃与溴化氢反应,分别生成溴代烷和1,1-溴代烷。反应遵从马氏规则,因为机理中涉及到碳阳离子,而由于碳阳离子会发生重排,因此有机会生成重排产物。机理是典型的亲电加成反应,首先是π电子进攻溴化氢,得到质子而生成碳阳离子和溴离子。之后溴离子进攻碳阳离子,生成溴代烷。机理如下:[20] [21] [22]

自由基加成[编辑]
在过氧化物的存在下,烯烃跟溴化氢的反应会是反马氏规则的,因为这涉及到自由基的生成[23]。例如:

醇的亲核取代反应[编辑]
醇可以跟氢溴酸反应,生成相应的溴代烷。但由于氢溴酸的酸性不够,所以需要用硫酸来增强酸性。这反应以三级醇最快,二级醇次之,一级醇最慢[24]。在反应体系中加入溴化钾,可以增加溴离子的浓度,并起了盐析的作用,是这反应的改良[25]。
醇可以在红磷与氢溴酸的体系中转化为溴代烷[26]。也可以直接用三溴化磷跟醇反应,生成溴代烷。
其他[编辑]
在阿佩尔反应中,用四溴化碳或溴作为卤原子源,可由醇制备出溴代烷[27]。伯醇、仲醇和多数叔醇都能顺利发生反应。
在汉斯狄克反应中,羧酸的银盐在无水的惰性溶剂如四氯化碳中与一分子溴回流,失去二氧化碳并形成比羧酸少一个碳的溴代烷[28][29][30][31] 。

汉斯狄克反应属于自由基机理。首先羧酸银与溴 (1) 快速反应而生成中间体 (2)。中间体在受热的情况下均裂为酰氧基自由基和溴自由基 (3),然后 RCOO· 分解放出二氧化碳,生成烷基自由基 R·,并重新与溴自由基结合为溴代烃 (5)。

同时,可以透过卤仿反应,将含乙酰基的有机化合物转变成溴仿[32]。

Haloform reaction scheme

首先,羰基的α-氢在碱性环境之下不断被溴取代[33],取代的过程重复三次,直到所有的氢原子都被换成溴原子。然后,氢氧根离子进攻羰基碳,离子离去。离子被碳上三个吸电子基团(溴)所稳定。最后一步为酸碱反应,从羧酸取得一质子,生成羧酸根离子和溴仿。其后羧酸根离子可以得到质子,重新生成羧酸。所生成的羧酸比原本的羰基化合物少一个碳[34]。反应的机理如下:



反应[编辑]
亲核取代反应[编辑]
溴的电负性比碳的强,因此碳原子上有部分正电荷,可以受亲核试剂的进攻。而溴是一个挺好的离去基团,使溴代烷相对上比较容易发生亲核取代反应,可以被氢氧根离子等各种亲核试剂进攻。例如,溴乙烷和氢氧根离子发生双分子亲核取代反应,经过五配位的过渡态后生成乙醇。

又如溴乙烷与乙酸钠反应,作为亲核试剂的乙酸根离子(乙酰氧基负离子)进攻,溴以溴离子的形式离去,生成乙酸乙酯:

由于位阻效应较小和所生成的一级碳阳离子不稳定,一级溴代烷通常以双分子亲核取代反应的反应机理进行反应。而三级溴代烷因为位阻较大,而且能生成较稳定的碳阳离子,所以通常以单分子亲核取代反应的机理而进行反应[35][36]。
值得注意的是溴代烷和两位负离子的反应。以亚硝酸根负离子做例子:一级溴代烷与其发生SN2反应,由于氮的亲核性强,产物为硝基烷。但如果是二级、三级溴代烷与其作SN1反应,由于碳阳离子跟亚硝酸根负离子上负电荷较为集中的氧有正负相吸,因此产物主要为亚硝酸酯[37]。
威廉姆逊合成反应[编辑]
醇羟基在碱性条件下去质子化,形成醇负离子,进攻溴代烷中带部分正电荷的碳。溴是离去基团,以溴离子的形式离开,形成醚键[38]。这是一个合成醚的重要反应。反应机理为双分子亲核取代反应,如下:

科尔贝腈合成[编辑]
氰离子进攻溴代烷,生成比原本溴代烷多一个碳的腈[39]。所生成的腈可以进一步反应为其他有机化合物(如水解成羧酸[40]、催化加氢合成胺[41][42]),因此这反应在有机合成上非常重要。

例如1-溴丁烷和氰化钠作用得到正戊腈和大约1%的异腈,此反应在二甲亚砜中进行得到93%产率[43]。
加布里尔伯胺合成反应[编辑]
邻苯二甲酰亚胺的钠盐或钾盐与一级溴代烷发生亲核取代反应,生成烷基邻苯二甲酰亚胺,再转换成一级胺[44][45][46][47]和副产物邻苯二甲酰肼[48]。此反应无法制备二级胺(仲胺),因为邻苯二甲酰亚胺的氮上只有一个氢原子,只能引入一个烷基。反应机理如下:

烷基化[编辑]
溴的电负性比碳大 (2.9 > 2.5)。因此,碳-溴键是极性共价键,其中的碳为亲电性的,可被亲核试剂进攻,从而增长碳链,因此溴代烷可以用作烷基化试剂。
酯、酮等含羰基的化合物可以在强碱的作用下失去质子,生成烯醇负离子[49][50][51][52]。而烯醇负离子可被视作亲核试剂,进攻溴代烷,生成α-烃基化的羰基化合物,这反应速率最快的为一级溴代烷和甲基溴[53]。

烯醇负离子的共振式

溴代烷更可以跟乙炔钠等炔化物反应,生成内部炔烃,加长碳链。除此之外,溴代烷可以参与乙酰乙酸酯合成[54]、丙二酸酯合成[55][56]等反应,这些反应的过程亦涉及到烷基化。以下是1,4-二溴丁烷进行分子内丙二酸酯合成(珀金脂环化合物合成)生成环烷羧酸的例子:
.png)
消除反应[编辑]
溴代烷可以发生消除反应,生成烯烃。而产物主要为柴瑟夫产物,即比较稳定的烯烃[57]。
双分子消除反应[编辑]
溴代烷在强碱的作用下失去一分子的溴化氢,生成烯烃。离去的溴和β氢必定处于反式共平面状态[58]。由于反应由一步完成(碱拉走质子(进攻β-氢)时与离去基同时离去),因此双分子消除反应与二种反应物浓度皆有关,在反应动力学上是属于二级反应。若果使用越强的碱,反应越快进行。1-溴-2-甲基丙烷和乙醇钾的反应机理如下:

Scheme 1. E2 reaction mechanism

单分子消除反应[编辑]
三级溴代烷在没有碱(溶剂解)的消除反应通常为单分子消除反应。第一步是溴代烷的溴以离去基团的形式离去,生成碳正离子。这一步的速率较慢,因此是速率控制步骤。第二步是溶剂分子夺取碳正离子β-氢,生成烯烃。由于反应的速率控制步骤是溴离去的一步,只与一个底物分子有关,因此反应是单分子过程,在反应动力学上是一级反应[42]。而由于反应中的碳阳离子有机会发生重排,因此单分子消除反应有机会生成重排产物。 以2-溴-2-甲基丙烷的单分子消除反应作例,反应机理如下:

单分子消除反应

与金属的反应[编辑]
格氏试剂的制备[编辑]

格氏试剂一般由溴代烷与金属镁在无水乙醚或四氢呋喃中直接反应而制备的[59]。由于格氏试剂非常活泼,因此制备时不能接触水、二氧化碳,或其他含有活泼氢的化合物(如醇),需要在惰性气体保护下制备格氏试剂。
格氏试剂是一个很好的亲核试剂,可以与不同的化合物反应(如醛、酮、二氧化碳等),增长有机化合物的碳链,在有机合成中非常重要[60]。
有机锂试剂的制备[编辑]

而由于有机锂试剂比格氏试剂更为活泼,所以制备的条件也要更为严格。
而二分子烃基锂与一分子卤化亚铜可生成二烃基铜(吉尔曼试剂)[62]:


偶联反应[编辑]

吉尔曼试剂与有机卤化物的化学反应方程式

在武兹反应里,两分子的溴代烃与钠反应,生成更长的碳链[64]:

其他的金属(如银等)都可参与反应[65]。邻二溴化物发生武兹反应,会生成烯烃;偕二溴化物反应,会生成炔烃;而(1,3), (1,4), (1,5),或(1,6)二溴化物发生这个反应,则会生成环状化合物[66]。而此反应虽然只限制于对称烷烃的合成,但其用处却是无可否认的,特别是在关很多小分子环(尤其是三元环)的时候相当有用。 比如二环丁烷就是通过1-溴-3-氯环丁烷和钠来合成的:

还原反应[编辑]
溴代烷可以被氢化铝锂还原成相应的烷烃[68][69],反应一般在乙醚或四氢呋喃中进行。例如溴辛烷能被还原成辛烷,产率为72%[70]。溴代烷的反应比碘代烷的慢(碘代烷的反应最快)。此反应中以还原一级溴代烷为最适合,所得产物发生构型转化,因此认为该反应是双分子亲核取代反应的机理。二级溴代烷也可用此法还原,三级溴代烷容易发生消除反应,不适合用这个方法。[71]
鉴定[编辑]
溴代烷与碘化钠的丙酮溶液反应会生成溴化钠。而由于溴化钠是不溶于丙酮的,会沉淀出来,因此可用这反应来区分溴代烷[72]。化学式如下:

同时,溴代烷(及其他卤代烷)在铜丝上燃烧可以产生绿色的火焰,这是鉴定卤素的简便方法[73]。
例子[编辑]
-
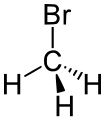 溴甲烷
溴甲烷 -
 溴仿
溴仿 -
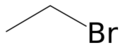 溴乙烷
溴乙烷


参见[编辑]
参考资料[编辑]
- ^ 卤代烷 (PDF). 太平洋化工资源网: p.20. [2021-09-01]. (原始内容存档 (PDF)于2021-08-31) (中文).
同一烃基卤烷的相对密度:碘烷 > 溴烷 > 氯烷。
- ^ E. Cheng, J. Chow, Y.F. Chow ,A. Kai , S.L. Lee, W.H.Wong. HKDSE Chemistry A Modern View Second Edition. : 42.18. ISBN 9789888242948 (英语).
- ^ Dagani, M. J.; Barda, H. J.; Benya, T. J.; Sanders, D. C., Bromine Compounds, Ullmann's Encyclopedia of Industrial Chemistry, Weinheim: Wiley-VCH, 2005, doi:10.1002/14356007.a04_405
- ^ Bromomethane. PubChem. [2021-09-01]. (原始内容存档于2013-06-19) (英语).
- ^ Gordon W. Gribble "The diversity of naturally occurring organobromine compounds" Chemical Society Reviews, 1999, volume 28, pages 335 – 346.doi:10.1039/a900201d
- ^ Rhoda A. Marshall, John T.G. Hamilton , M.J. Dring, D.B. Harper. Do vesicle cells of the red alga Asparagopsis (Falkenbergia stage) play a role in bromocarbon production? Chemosphere 52 (2003) 471–475.
- ^ Gordon, H. Laing. Sir James Young Simpson and Chloroform (1811-1870). The Minerva Group, Inc. 2002-11: 106–109 [11 November 2011]. ISBN 978-1-4102-0291-8. (原始内容存档于2020-08-19).
- ^ 陈炳圣. 《萬物簡史》. 源桦. 2007. ISBN 986828421X.
- ^ Aug. Kekulé. Über die s. g. gepaarten Verbindungen und die Theorie der mehratomigen Radicale. Annalen der Chemie und Pharmacie. 1857, 104 (2): 129–150. doi:10.1002/jlac.18571040202.
- ^ CHAPTER 1 INTRODUCTION TO ORGANIC CHEMISTRY (PDF). 南伊利诺斯大学艾德华兹维尔分校. [2021-09-01]. (原始内容存档 (PDF)于2020-10-30) (英语).
- ^ Additions to Alkenes: Regiochemistry. [2021-09-01]. (原始内容存档于2020-02-19).
- ^ Illustrated Glossary of Organic Chemistry. 加利福尼亚大学洛杉矶分校. [2021-09-01]. (原始内容存档于2021-09-01) (英语).
Peroxide effect: The change in regioselectivity of the addition of HBr to an alkene or alkyne in the presence of a peroxide. The regioselectivity for the addition reactions of other electrophiles such as HCl and H3O+ are not altered in the presence of a peroxide.
- ^ molbase. 2-溴丙烷. 上海摩库数据技术有限公司. [2021-07-28]. (原始内容存档于2021-07-28) (中文).
- ^ 有机化合物命名原则2017 (页面存档备份,存于互联网档案馆). 中国化学会. [2018-1-17]
- ^ 卤代烷 (PDF). 太平洋化工资源网: p.5. [2021-08-31]. (原始内容存档 (PDF)于2021-08-31) (中文).
把卤烷看作是烷基和卤素结合而成的化合物而命名,称为某烷某卤。
- ^ Dr. Ian Hunt,. Chapter 4: Alcohols and Alkyl Halides. [2021-07-28]. (原始内容存档于2021-07-28) (英语).
Note that it contains three distinct type of steps
- ^ 国际纯化学和应用化学联合会,化学术语概略,第二版。(金皮书)(1997)。在线校正版: (2006–) "homolysis (homolytic)"。doi:10.1351/goldbook.H02851
- ^ E. Cheng, J. Chow, Y.F. Chow ,A. Kai , S.L. Lee, W.H.Wong. HKDSE Chemistry A Modern View Second Edition. : 44.4. ISBN 9789888242948 (英语).
- ^ THE REACTION BETWEEN SYMMETRICAL ALKENES AND BROMINE. 2000 [2021-07-28]. (原始内容存档于2021-12-08) (英语).
- ^ Solomons, T.W. Graham; Fryhle, Craig B., Organic Chemistry 8th, Wiley, 2003, ISBN 0471417998
- ^ Smith, Janice G., Organic Chemistry 2nd, McGraw-Hill, 2007, ISBN 0073327492
- ^ P.J. Kropp, K.A. Dans, S.D. Crawford, M.W. Tubergen, K.D. Kepler, S.L Craig, and V.P. Wilson, Surface-mediated reactions. 1. Hydrohalogenation of alkenes and alkynes, J. Am. Chem. Soc., 1990, 112 (112): 7433–7434, doi:10.1021/ja00176a075.
- ^ Streiwieser, A.; Heathcock, C.H.; Kosower, E.M. 11.6.G. Alkenes: Reactions: Free Radical Additions. Introduction to Organic Chemistry 4th. New York: Macmillan. 1992: 288.
- ^ 邢其毅; 裴坚; 裴伟伟; 徐瑞秋. 醇羥基轉換爲鹵原子的反應. 基礎有機化學. 北京大学出版社. : 285. ISBN 9787301272121 (中文).
- ^ 张永华, 丁辰元, 张国玺. 叔丁基溴合成方法的改进 (页面存档备份,存于互联网档案馆)[J]. 首都师范大学学报:自然科学版, 1989(2):21-23.
- ^ 溴丙烷. molbase. 上海摩库数据技术有限公司. [2021-07-28]. (原始内容存档于2021-07-28) (中文).
另外,在红磷存在下,正丙醇与溴反应也可制备溴丙烷。
- ^ Rolf Appel. Tertiary Phosphane/Tetrachloromethane, a Versatile Reagent for Chlorination, Dehydration, and P-N Linkage. Angewandte Chemie International Edition in English. 1975, 14 (12): 801–811. doi:10.1002/anie.197508011.
- ^ Cläre Hunsdiecker, et al. U.S. Patent # 2,176,181.
- ^ Heinz Hunsdiecker; Cläre Hunsdiecker. Über den Abbau der Salze aliphatischer Säuren durch Brom. Ber. 1942, 75: 291. doi:10.1002/cber.19420750309.
- ^ Borodin, A. Ueber Bromvaleriansäure und Brombuttersäure. Ann. 1861, 119: 121. doi:10.1002/jlac.18611190113.
- ^ Allen, C. F. H.; Wilson, C. V. (1955). "Methyl 5-bromovalerate". Org. Synth.; Coll. Vol. 3: 578.
- ^ Chakrabartty, in Trahanovsky, Oxidation in Organic Chemistry, pp 343-370, Academic Press, New York, 1978
- ^ March, Jerry; Smith, Michael B. Knipe, A.C. , 编. March's Advanced Organic Chemistry Reactions, Mechanisms, and Structure. 6th. Hoboken: John Wiley & Sons. 2007: 484. ISBN 9780470084946.
- ^ Reynold C. Fuson and Benton A. Bull. The Haloform Reaction. Chemical Reviews. 1934, 15 (3): 275–309. doi:10.1021/cr60052a001.
- ^ 8.4 Comparison and Competition Between SN1, SN2, E1 and E2. Pressbooks. [2021-07-28]. (原始内容存档于2021-07-28) (英语).
- ^ SN1 and SN2 Reactions (pdf). [2021-07-28]. (原始内容存档 (PDF)于2019-05-12) (英语).
- ^ 邢其毅; 裴坚; 裴伟伟; 徐瑞秋. 影響鹵代烴親核取代反應的因素. 基礎有機化學. 北京大学出版社. : 239. ISBN 9787301272121 (中文).
- ^ Theory of Aetherification. Philosophical Magazine. 1850, 37: 350–356.)
- ^ Organikum, 22. Edition (German), Wiley-VCH, Weinheim, 2004, ISBN 3-527-31148-3
- ^ 羧酸的制备——腈水解 (页面存档备份,存于互联网档案馆). 北京中医药大学远程教育学院
- ^ Nishimura, Shigeo. Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis 1st. New York: Wiley-Interscience. 2001: 254–277. ISBN 9780471396987.
- ^ 42.0 42.1 March, Jerry (1985). Advanced Organic Chemistry, Reactions, Mechanisms and Structure, third Edition, John Wiley & Sons. ISBN 0-471-85472-7.
- ^ L. Friedman, Harold Shechter. Preparation of Nitriles from Halides and Sodium Cyanide. An Advantageous Nucleophilic Displacement in Dimethyl Sulfoxide. Journal of Organic Chemistry. 1960, 25: 877–879. doi:10.1021/jo01076a001.
- ^ Gabriel, S. Ueber eine Darstellung primärer Amine aus den entsprechenden Halogenverbindungen. Ber. 1887, 20: 2224 [2021-09-15]. (原始内容存档于2020-04-10).
- ^ Sheehan, J. C.; Bolhofer, V. A. An Improved Procedure for the Condensation of Potassium Phthalimide with Organic Halides. J. Am. Chem. Soc. 1950, 72: 2786. doi:10.1021/ja01162a527.
- ^ Gibson, M.S.; Bradshaw, R.W. The Gabriel Synthesis of Primary Amines. Angew. Chem. Int. Ed. Engl. 1968, 7: 919. doi:10.1002/anie.196809191.
- ^ Mitsunobu, O. Comp. Org. Syn. 1991, 6, 79-85.(综述)
- ^ Khan, M. N. Suggested Improvement in the Ing-Manske Procedure and Gabriel Synthesis of Primary Amines: Kinetic Study on Alkaline Hydrolysis of N-Phthaloylglycine and Acid Hydrolysis of N-(o-Carboxybenzoyl)glycine in Aqueous Organic Solvents. J. Org. Chem. 1996, 61, 8063-8068.
- ^ Stolz, Daniel; Kazmaier, Uli. Metal Enolates as Synthons in Organic Chemistry. PATai's Chemistry of Functional Groups. 2010. ISBN 9780470682531. doi:10.1002/9780470682531.pat0423.
- ^ Hart, David J.; Ha, Deok Chan. The ester enolate-imine condensation route to .beta.-lactams. Chemical Reviews. 1989, 89 (7): 1447–1465. doi:10.1021/cr00097a003.
- ^ Wu, George; Huang, Mingsheng. Organolithium Reagents in Pharmaceutical Asymmetric Processes. Chemical Reviews. 2006, 106 (7): 2596–2616. PMID 16836294. doi:10.1021/cr040694k.
- ^ Curti, Claudio; Battistini, Lucia; Sartori, Andrea; Zanardi, Franca. New Developments of the Principle of Vinylogy as Applied to π-Extended Enolate-Type Donor Systems. Chemical Reviews. 2020, 120 (5): 2448–2612. PMID 32040305. doi:10.1021/acs.chemrev.9b00481
 .
.
- ^ Alkylation of enolates. ChemistryScore. [2021-07-28]. (原始内容存档于2021-07-28) (英语).
In the first step, the base (LDA) removes a proton from the α carbon to produce an enolate. In the second step, the enolate function as a nucleophile and attacks the alkyl halide, displacing the halide as a good leaving group. The alkylation product is formed by the SN2 reaction. This reaction works best with methyl and primary alkyl halides. Hindered alkyl halides and those with halides bonded to sp² hybridized carbon do not undergo substitution.
- ^ Smith, Janice Gorzynski. Organic Chemistry: Second Ed. 2008. pp 905-906
- ^ House, Herbert O. Modern Synthetic Reactions. Menlo Park, CA.: W. A. Benjamin. 1972. ISBN 0-8053-4501-9.
- ^ Malonic Ester Synthesis. Organic Chemistry Portal. [2007-10-26]. (原始内容存档于2018-07-29).
- ^ Saytzeff, Alexander. Zur Kenntniss der Reihenfolge der Analgerung und Ausscheidung der Jodwasserstoffelemente in organischen Verbindungen. Justus Liebigs Annalen der Chemie. 1875, 179 (3): 296–301. doi:10.1002/jlac.18751790304.
- ^ David R. Klein. 有機化學天堂秘笈. Taiwan: 天下远见出版股份有限公司. Oct 19, 2007: 302~309. ISBN 978-986-216-008-4 (中文(香港)).
- ^ Smith, M. B.; March, J. Advanced Organic Chemistry, 5th ed.; John-Wiley & Sons: New York, 2001. ISBN 0-471-58589-0 - 包含格氏试剂的若干反应。
- ^ Kürti, L.; Czakó, B. Storategic Applications of Named Reactions in Organic Chemistry; Elsevier: Amsterdam, 2005; pp 188–189. ISBN 0-12-429785-4
- ^ Stent, M. Generation of a Highly Basic and Nucleophilic Organolithium; Isopropyllithium. Synthetic Pages. 2002, (195) [2021-07-29]. (原始内容存档于2016-03-10).
- ^ Modern Organocopper Chemistry, N. Krause Ed. Wiley-VCH, 2002.
- ^ J. F. Normant. Organocopper(I) Compounds and Organocuprates in Synthesis. Synthesis. 1972, 1972 (02): 63–80. doi:10.1055/s-1972-21833.
- ^ Adolphe Wurtz. Sur une nouvelle classe de radicaux organiques. Annales de chimie et de physique. 1855, 44: 275–312 [2021-07-28]. (原始内容存档于2020-04-10).
- ^ March Advanced Organic Chemistry 5th edition p.535
- ^ Gary M. Lampman and James C. Aumiller "Bicyclo[1.1.0]butane" Organic Syntheses, 1971, volume 51, pp 55-9. doi:10.15227/orgsyn.051.0055 Aditya Krishna "Dihalides Quartz"
- ^ 邢其毅; 裴坚; 裴伟伟; 徐瑞秋. 鹵代烴與金屬的反應. 基礎有機化學. 北京大学出版社. : 265. ISBN 9787301272121 (中文).
- ^ Johnson, J. Enoch; Blizzard, Ronald H.; Carhart, Homer W. Hydrogenolysis of alkyl halides by lithium aluminum hydride.. Journal of the American Chemical Society. 1948, 70 (11): 3664. PMID 18121883. doi:10.1021/ja01191a035.
- ^ Krishnamurthy, S.; Brown, Herbert C. Selective reductions. 28. The fast reaction of lithium aluminum hydride with alkyl halides in THF. A reappraisal of the scope of the reaction. The Journal of Organic Chemistry. 1982, 47: 276. doi:10.1021/jo00341a018.
- ^ 邢其毅; 裴坚; 裴伟伟; 徐瑞秋. 鹵代烴的還原. 基礎有機化學. 北京大学出版社. : 259. ISBN 9787301272121 (中文).
- ^ Carruthers, W. Some modern methods of organic synthesis. Cambridge University Press. 2004: 470. ISBN 0521311179.
- ^ 邢其毅; 裴坚; 裴伟伟; 徐瑞秋. 鹵代烴親核取代反應的應用. 基礎有機化學. 北京大学出版社. : 248-249. ISBN 9787301272121 (中文).
- ^ 卤烷的物理性质 (PDF). 太平洋化工资源网: p.20. [2021-09-01]. (原始内容存档 (PDF)于2021-08-31) (中文).
- ^ 三溴甲烷. chemicalbook. [2021-07-28]. (原始内容存档于2021-07-28) (中文).
另外可作为消毒剂、镇痛剂、致冷剂、防火化学品,测定分子量的溶剂以及有机合成的中间体。
- ^ 溴乙烷. chemicalbook. [2021-07-28]. (原始内容存档于2021-07-28).
用于医药、农药、染料工业,是有机合成中的乙基化剂,也用作致冷剂和有机溶剂